
























SMA Hastalığı Nedir? Neden Olur? Belirtileri ve Tedavisi
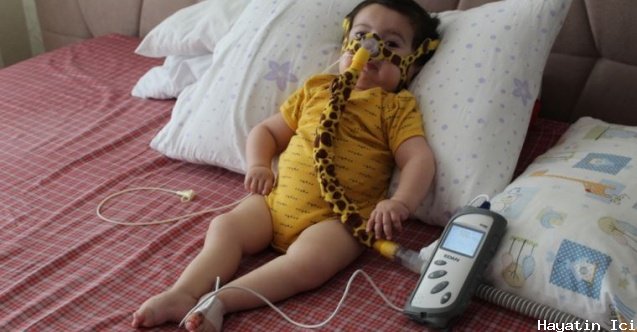

Spinal müsküler atrofi (SMA), omurilikte alt motor nöronlar veya ön boynuz hücreleri olarak adlandırılan sinir hücrelerinin kaybı ile karakterize bir grup kalıtsal nöromüsküler bozukluktur. Alt motor nöronlar beyin sapından veya omurilikten kaynaklanır ve beyinde bulunan üst motor nöronlardan gelen sinir uyarılarını kontrol ettikleri kaslara iletir.
SMA Hastalığı Hakkında Bilmeniz Gereken Her Şey

Alt motor nöronların kaybı, tipik olarak omuzlar, kalçalar ve sırt gibi vücudun gövdesine en yakın kaslarda (proksimal kaslar) daha belirgin olan ilerleyici kas zayıflığına, kas erimesine (atrofi) ve düşük kas tonusuna (hipotoni) yol açar. . Bununla birlikte, beslenme, yutma ve nefes alma ile ilgili kasları kontrol edenler de dahil olmak üzere, gönüllü kasların çoğunu kontrol eden nöronlar etkilenebilir.
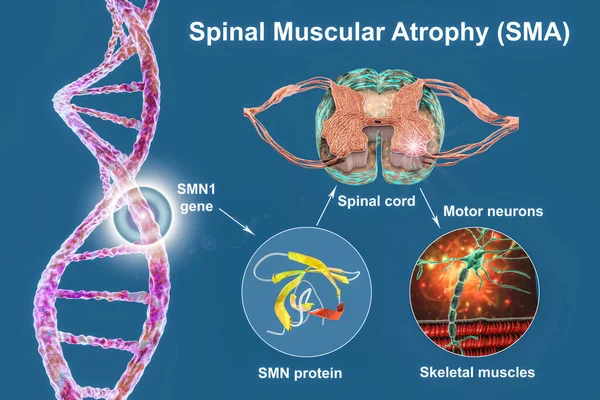
SMA, semptomların başlama yaşına ve elde edilen maksimum motor fonksiyona bağlı olarak alt tiplere (SMA tipleri 0 ila 4) ayrılır; daha düşük bir sayı, daha genç bir başlangıç yaşını ve daha şiddetli hastalığı temsil eder. SMA, otozomal resesif bir genetik bozukluk olarak kalıtılır ve hayatta kalan motor nöron 1 ( SMN1 ) genindeki mutasyonlarla ilişkilidir. SMN1 uzun kol (q) bölgesinde 5. kromozom üzerinde yer alır. Bu nedenle, bir SMN1 gen delesyonu olan SMA, genellikle 5q SMA olarak adlandırılır ve bu SMA formunu diğer SMA genetik formlarından ayırır.
Yenidoğan taraması, SMA'lı bebeklerin erken teşhisini ve dolayısıyla tedavinin erken uygulanmasını kolaylaştırır. SMA yenidoğan taraması ile tanımlanan bebekler, doğrulayıcı testler, tedavilerin tartışılması ve bakım için acilen sevk edilir. Semptomların başlangıcından önce erken tedavi en iyi sonuçları sağlar.
SMA'nın yönetimi daha önce semptom yönetimi ve destekleyici bakım etrafında odaklanmış olsa da, 2016'dan beri hastalığın seyrini iyileştirebilecek tedaviler (hastalığı modifiye edici tedaviler) ortaya çıkmış ve umut verici sonuçlar vermiştir. Şu anda üç SMN geliştirici tedavi ABD Gıda ve İlaç Dairesi (FDA) onayına sahiptir.
SMA Hastalığı İşaretler ve Belirtileri
SMA'nın belirti ve semptomları, düşük motor nöron kaybının bir sonucudur. Alt motor nöron hastalığının özellikleri arasında kas zayıflığı ve atrofisi, hipotoni, reflekslerin azalması veya olmaması (hipo- veya arefleksi) ve kas liflerinin seğirmesi (fasikülasyonlar) bulunur. SMA bir hastalık spektrumu olmasına rağmen, beş alt tip, semptomların başlama yaşlarına ve ulaşılan maksimum motor fonksiyona göre belirlenir. SMA için bu sınıflandırma, genetik testlerin mevcudiyetinden ve hastalık modifiye edici tedavilerin mevcudiyetinden önce oluşturulmuştur.

Prenatal SMA olarak da bilinen SMA tip 0, hastalığın en şiddetli şeklidir ve doğumdan önce gelişir. İlk işaret, geç gebelik sırasında fetal harekette bir azalma veya kayıp olabilir. SMA tip 0 semptomları doğumda belirgindir ve şiddetli halsizlik ve hipotoni içerir. Ayrıca eklem deformitesi ve daralması (kasılmalar) ve doğuştan kalp kusurları sık görülür. Sonuç olarak, bebekler gelişimsel motor kilometre taşlarına ulaşamazlar. Şiddetli solunum kas güçsüzlüğü nedeniyle, etkilenen bebekler genellikle yaşamın ilk ayında hızla solunum yetmezliğine ilerler.
İnfantil SMA veya Werdnig-Hoffmann hastalığı olarak da bilinen SMA tip 1, SMA ile doğan bebeklerin yaklaşık %60'ını etkileyen en yaygın SMA tipidir ve aynı zamanda hastalığın ciddi bir şeklidir. SMA tip 1 olan bebekler genellikle doğumda normal görünürler ancak 6 aylıktan önce ciddi güçsüzlük yaşarlar. Gelişimsel olarak bağımsız oturmayı başaramazlar ve çok az gelişimsel motor kilometre taşına ulaşabilirler. Daha düşük motor nöron kaybı nedeniyle, etkilenen bebeklerin emme ve yutma refleksleri ve solunum kasları zayıflığı vardır. Tarihsel olarak müdahale olmadan, etkilenen çocuklar ilerleyici solunum kas zayıflığı ve solunum yetmezliği nedeniyle iki yaşından önce ölürler.
Ara SMA veya Dubowitz hastalığı olarak da bilinen SMA tip 2, SMA ile doğan bebeklerin yaklaşık %30'unu oluşturur. Hastalık genellikle 6 ila 18 aylıkken kendini gösterir. Etkilenen çocuklar, gelişimlerinin bir noktasında bağımsız olarak oturabilirler. Bununla birlikte, bu yetenek genellikle ergenlik çağının ortalarında veya sonrasında kaybolur ve etkilenen bireyler asla bağımsız ayakta durma ve yürümeyi başaramazlar. Ek ilişkili semptomlar arasında yutma güçlüğü (disfaji) ve solunum güçlükleri yer alır. Parmakların titremesi (titremesi) de yaygındır. Ayrıca omurgayı destekleyen kasların zayıf olması da omurganın eğriliğine (skolyoz) yol açar. Tarihsel olarak, SMA tip 2 hastalarında yaşam beklentisi azalır, ancak çoğu yetişkinliğe ulaşır.
Juvenil SMA veya Kugelberg-Welander hastalığı olarak da bilinen SMA tip 3, SMA ile doğan bebeklerin yaklaşık %10'unu oluşturur. Başlangıç yaşı değişkendir ve 18 ay kadar erken veya gençlik yılları kadar geç olabilir. Etkilenen bireylerin kalça ve bacak zayıflığı olmasına ve sıklıkla düşebilmelerine rağmen, gelişimlerinin bir noktasında bağımsız olarak yürüyebilirler. Bununla birlikte, büyüdükçe ve hastalığın ilerlemesiyle birlikte yürüme ve ayakta durma yeteneği kaybolabilir ve birçoğu tekerlekli sandalyeye bağımlı hale gelir. Uzun vadeli prognoz, çocukken kazanılan motor fonksiyonun derecesine bağlıdır ve solunum kas zayıflığı tipik olarak hafiftir veya yoktur. SMA tip 3, normal bir yaşam beklentisi ile ilişkilidir.
Geç başlangıçlı SMA olarak da bilinen SMA tip 4, SMA'lı kişilerin %1'inden azında görülür. Semptomlar diğer alt tiplere göre daha az şiddetlidir ve başlangıç tipik olarak yetişkinlikte ve en yaygın olarak 35 yaşından sonra ortaya çıkar. Tüm motor gelişimsel dönüm noktalarına ulaşılır ve SMA tip 4'lü çoğu kişi yaşamları boyunca yürüyebilir. SMA tip 4 hastaları normal bir yaşam beklentisine sahiptir.
SMA Hastalığı Nedenleri
SMA'ya , hayatta kalma motor nöron (SMN) olarak bilinen bir proteini kodlayan SMN1 genindeki delesyon veya mutasyon neden olur . Bu protein, motor nöronların işleyişinde ve korunmasında önemli bir rol oynar. Etkilenen bireylerin yaklaşık %95-98'inde SMN1 geninde delesyonlar ve %2-5'inde SMN1 geninde SMN proteininin üretiminin azalmasına neden olan bir nokta mutasyonu vardır.
SMN2 geni, SMN1'in bir paralogudur ve ayrıca SMN proteinini kodlar ve SMN1 geninin kaybını kısmen telafi edebilir . Bununla birlikte, SMN2 geni tarafından üretilen çoğu SMN proteini işlevsel değildir, bu da SMN2 geninin SMN1 geninin kaybını yalnızca kısmen telafi edebileceği anlamına gelir . Bu nedenle, SMN2 geninin daha fazla kopyasına sahip olan SMA'lı bir birey, daha işlevsel SMN proteini üretecek ve SMN1 geninin kaybını daha iyi telafi edebilecek ve dolayısıyla daha az şiddetli hastalığa yol açabilecektir. Genellikle, SMN2'nin daha fazla kopyasıistisnalar olsa da, daha hafif SMA hastalığı ile ilişkilidir.
SMA, otozomal resesif bir modelde kalıtılır. Çekinik genetik bozukluklar, bir birey her bir ebeveynden hastalık için bir patojenik gen varyantı aldığında ortaya çıkar. Bir kişi hastalık için bir normal gen ve bir varyant gen alırsa, kişi hastalık için taşıyıcı olur, ancak genellikle semptom göstermez. Taşıyıcı iki ebeveynin her ikisinin de varyant geni geçirme ve bu nedenle etkilenen bir çocuğa sahip olma riski her hamilelikte %25'tir. Ebeveynler gibi taşıyıcı bir çocuğa sahip olma riski her hamilelikte %50'dir. Bir çocuğun her iki ebeveynden de normal genleri alma şansı %25'tir. Erkekler ve kadınlar için risk aynıdır.
Yakın kan akrabası (akraba) olan ebeveynlerin aynı zararlı gen varyantına sahip olma ve dolayısıyla etkilenmiş bir çocuğa sahip olma olasılığı daha yüksektir.
SMA Hastalığından Etkilenen Popülasyonlar
SMA insidansı yaklaşık 10.000 canlı doğumda 1'dir. SMA, kadınları ve erkekleri eşit olarak etkiler.
İlgili Bozukluklar
Aşağıdaki bozuklukların belirtileri SMA'nınkilere benzer olabilir. Karşılaştırmalar ayırıcı tanı için faydalı olabilir.
SMN1 genindeki anormalliklerin neden olduğu biçimlerin ötesinde SMA'nın başka biçimleri de vardır . SMA'nın bu diğer formları, tercihen vücudun belirli kısımlarını etkilemelerine ve diğer semptomlarla ilişkili olmalarına rağmen, alt motor nöronları etkiler. Bu SMA tipleri özellikle skapuloperoneal SMA, pontoserebellar hipoplazili SMA, artrogripozlu X'e bağlı infantil SMA, solunum sıkıntısı tip I (SMARD1) olan SMA, konjenital distal SMA, distal SMA-V/CMT2d ve Finkel tip SMA'yı içerir.
Konjenital miyastenik sendromlar, kas ve sinir iletişiminin (nöromüsküler iletim) genetik kusurlarından kaynaklanır. Bu durumlar genellikle bebeklerde ortaya çıkar, ancak yetişkinlikte belirgin hale gelebilir. İlişkili özelliklerin şiddeti kişiden kişiye değişebilir. Semptomlar, beslenme güçlüklerini, ani spontan solunum yokluğu epizodlarını (apne), büyümede ve beklenen oranda kilo almada (gelişmede başarısızlık), kaslarda zayıflık ve yorgunluk, göz kaslarında güçsüzlük veya felç (oftalmopleji) ve diğer anormallikleri içerebilir.
Konjenital miyopatiler, kasları etkileyen (miyopati) ve genellikle doğumda ortaya çıkan güçsüzlük ve hipotoni ile karakterize kalıtsal bir hastalık grubudur. Konjenital miyopatilerin birkaç farklı alt tipi vardır ve bunların çoğuna spesifik genlerdeki patojenik varyantlar neden olur. Semptomların şiddeti ve başlangıcı, mikroskop altında hücresel özellikler ve prognoz açısından farklılık gösterirler. Semptomlar doğumdan itibaren mevcut olabilir veya bebeklik ve çocukluk boyunca yavaş ilerleyebilir, ancak bu bozukluklar yetişkinlikte tipik olarak daha şiddetli olmaz. (Bu bozukluklar hakkında daha fazla bilgi için Nadir Hastalıklar Veritabanında arama teriminiz olarak “Konjenital Miyopati”yi seçin.)
Konjenital müsküler distrofi (CMD), doğumda veya bebeklik döneminde erken dönemde ortaya çıkan başka bir genetik kas hastalıkları grubu için genel bir terimdir. CMD'ler genellikle hipotoni, ilerleyici kas zayıflığı ve atrofi, kontraktürler, spinal sertlik ve motor kilometre taşlarına ulaşmada gecikmeler ile karakterizedir. Bazı hastalarda beslenme güçlüğü ve solunum komplikasyonları gelişebilir. Kas zayıflığı düzelebilir, sabit kalabilir veya kötüleşebilir. Bazı CMD formları, yapısal beyin anormallikleri ve zihinsel engellilikle ilişkilendirilebilir. Bu bozuklukların şiddeti, spesifik semptomları ve ilerlemesi büyük ölçüde değişir. Duchenne ve Becker müsküler distrofileri, genellikle ayrı olarak sınıflandırılan diğer iki tür müsküler distrofidir.
SMA Hastalığı Teşhis
Açıklanamayan zayıflığı ve hipotonisi olan, gözleri parlak ve sosyal olarak ilgi çekici görünen bir bebek gibi SMA'dan şüphelenilen bir hastanın değerlendirilmesi, tam bir hasta öyküsü ve fizik muayene ile başlar. Klinik değerlendirme, alt motor nöron hastalığı belirtileri gösteriyorsa (İşaretler ve Semptomlar bölümüne bakınız) ve SMA öneriyorsa, tanı, SMN1 genindeki patojenik varyantları saptamak için yapılan genetik testlerle doğrulanır ve SMN1'in hiçbir kopyası yoksa , refleks testi ile doğrulanır. SMN2 kopya numarası yarıştırılmalıdır. Hasta semptomatikse ve SMN1'in bir kopyası tanımlanmışsa, olası bir SMN1 nokta mutasyonunu değerlendirmek için gen dizi analizi yapılmalıdır .
SMA'yı teşhis etmek için başka hiçbir teste gerek yoktur, ancak benzer bir klinik sunuma sahip olabilecek diğer durumları dışlamak için başlangıçta ek testler yapılabilir. Bu, diğer hastalıklarla ilişkili genetik testleri, metabolik veya biyokimyasal testleri veya elektrik sinyallerinin sinirlerden kaslara iletiminin değerlendirilmesini (elektromiyografi; EMG) içerebilir. Yukarıdaki testler bir tanı ortaya koymadığında kas biyopsisi düşünülebilir.
SMA için yenidoğan taraması Amerika Birleşik Devletleri'nde uygulanmaktadır. Ocak 2021 itibariyle, ABD'de doğan tüm bebeklerin %86'sını temsil eden 39 eyalette SMA taraması Yenidoğan taraması, SMA'lı bebeklerin erken tanımlanmasını ve dolayısıyla tedavinin erken uygulanmasını kolaylaştırır. SMA yenidoğan taraması ile tanımlanan bebekler, doğrulayıcı testler, tedavilerin tartışılması ve bakım için acilen sevk edilir. Semptomların başlangıcından önce erken tedavi en iyi sonuçları sağlar. Yenidoğan taraması, SMN1 geninde nokta mutasyonu olması nedeniyle SMA'lı bebeklerin %3-5'ini belirlemeyecektir . Bu bebekler semptomlar geliştirmeye ilerleyecek ve hızlı tanı ve tedavi gerektirecektir.
SMA için taşıyıcı testi, SMN1 geninin kopya sayısının belirlendiği bir moleküler genetik test kullanılarak da mevcuttur. Amerikan Kadın Hastalıkları ve Doğum Uzmanları Koleji, hamileliği düşünen veya şu anda hamile olan tüm kadınlara SMA için taşıyıcı taraması yapılmasını önermektedir.
SMA Hastalığı Standart Terapiler
SMA tedavisi multidisipliner bir ekip yaklaşımı gerektirir ve özellikle nörologları, tıbbi genetikçileri, fizyoterapistleri, konuşma patologlarını, göğüs hastalıkları uzmanlarını, solunum terapistlerini, tıbbi sosyal hizmet uzmanlarını, beslenme uzmanlarını, psikologları ve uzman hemşireleri içermelidir. SMA yönetiminin iki ana bileşeni vardır: hastalığın ilerlemesini yavaşlatan tedavi (hastalığı değiştiren tedavi) ve semptomları yönetmeye yardımcı olan ve yaşam kalitesini iyileştiren tedavi (destekleyici tedavi). Etkilenen bireyler ve aileleri için genetik danışmanlık önerilir.
Semptomatik tedavi
SMA'nın semptomatik yönetimi, fizik tedavi, mesleki terapi, solunum fonksiyonunun izlenmesi ve klinik olarak belirtildiği şekilde müdahale edilmesi, beslenme durumunun izlenmesi ve müdahalesi, omurga eğriliğinin izlenmesi ve müdahalesi ve gerektiğinde ortez ve adaptif ekipmanın kullanımını içerir. SMA tip 1 için solunum desteği (6 aylıktan önce semptomatik olan bebekler), hipoventilasyonu yönetmek için BiPAP (iki seviyeli pozitif hava yolu basıncı) adı verilen solunum desteğini ve zayıf öksürüğü desteklemek için mekanik bir hava üfleme cihazını içerir. Destekleyici yönetimin konforu ve yaşam beklentisini arttırdığı gösterilmiştir. Hastalığın başlarında, etkilenen bazı bebekler sadece geceleri ventilasyon desteğine ihtiyaç duyabilir. İlerleyici solunum yetmezliği olan çocuklar nefes almak için daha invaziv müdahaleler gerektirebilir. örneğin boyundan bir solunum tüpünün cerrahi olarak yerleştirilmesi (trakeostomi). Disfajisi olan bebekler ve çocuklar için beslenme desteği, beslenmeyi güvenli bir şekilde sağlamak için gastrostomi tüpü yerleştirilmesini gerektirebilir. SMA'lı çocuklar ayrıca skolyoz ve/veya kalça çıkığı gibi kas-iskelet sistemi sorunları için cerrahi müdahale gerektirebilir.
Hastalık değiştirici tedavi
Araştırma çabaları, SMA'nın seyrini iyileştirebilecek tedavilere yol açmıştır. İlk hastalığı modifiye edici tedavi, 2016 yılında ABD Gıda ve İlaç Dairesi (FDA) tarafından onaylandı. Bu tedaviler, özellikle gelişimsel motor kilometre taşı başarısı ve tedavi edilen bireylerde daha iyi hayatta kalma gibi umut verici sonuçlar göstermiştir. Bu tedavilerin etkisi araştırılırken, bu tedavilerin tedavi olmadığını unutmayın.
2016 yılında, nusinersen (Spinraza), FDA tarafından SMA'lı çocukları ve yetişkinleri tedavi eden ilk ilaç olarak onaylandı. Nusinersen, omuriliği çevreleyen sıvıya uygulanan bir enjeksiyondur (intratekal uygulama). Nusinersen, daha tam uzunlukta ve fonksiyonel SMN proteini üretilecek şekilde SMN2 gen ürününün, mRNA'nın eklenmesini modifiye ederek etki eder.
2019'da FDA, SMA'lı iki yaşından küçük çocukların tedavisi için onasemnogen abeparvovec-xioi'yi (Zolgensma) onayladı. Onasemnogene abeparvovec-xioi, insan SMN1 geninin tamamen işlevsel bir kopyasını bir viral vektör, AAV9 aracılığıyla hedef motor nöron hücrelerine ileten bir gen tedavisidir . İlacın bir kerelik intravenöz uygulaması, motor nöronlar dahil tüm hücrelerde SMN proteininin artmasına neden olur.

2020'de FDA, iki aylık ve daha büyük hastaları SMA ile tedavi etmek için risdiplamı (Evrysdi) onayladı. Risdiplam, SMA tedavisi için onaylanmış ilk oral yoldan verilen ilaçtır. Ayrıca SMN2 mRNA'nın eklenmesini modifiye ederek SMN proteininin artmasına neden olan bir etki mekanizmasına sahiptir .





 : belirtiler, nedenler, tedaviler")






")
















