SMS sendromu nedir? Belirti ve tedavi yöntemleri nelerdir?
SMS sendromu nedir? Belirti ve tedavi yöntemleri nelerdir?
- SAĞLIK
- Sat, 21 May 2022 19:28:44
- Sat, 21 May 2022 19:28:44
Smith-Magenis sendromu ( SMS ), vücudun çoklu organ sistemlerini etkileyen karmaşık bir gelişimsel bozukluktur. Bozukluk, doğumda ( doğuştan ) mevcut olan bir anormallik modelinin yanı sıra davranışsal ve bilişsel problemlerle karakterizedir. Yaygın semptomlar arasında belirgin yüz özellikleri, iskelet bozuklukları, değişen derecelerde zihinsel yetersizlik, konuşma ve motor gecikmeler, uyku bozuklukları ve kendine zarar verme veya dikkat çekme davranışları yer alır.
SMS Sendromu Nedir? Neden Olur? Belirtileri ve Tedavisi
Her hastada mevcut olan spesifik semptomlar, bir bireyden diğerine önemli ölçüde değişebilir. Vakaların yaklaşık %90'ı, kromozomun bir kısmı eksik veya silindiğinde ( monozomik ) ortaya çıkar. 17p11.2 kromozomundaki bu silinmiş kısım bozukluğun gelişiminde önemli bir rol oynadığına inanılan gen RAI1'i içerir.. Kalan durumlarda, kromozom 17'de silinmiş materyal yoktur; bu vakalara RAI1 genindeki mutasyonlar neden olur .
Silinen segmentteki diğer genler de sendromdaki değişken özelliklerde rol oynayabilir, ancak SMS gelişiminde ne kadar önemli bir rol oynadıkları tam olarak anlaşılmamıştır. Kalan durumlarda, kromozom 17'de silinmiş materyal yoktur; bu vakalara RAI1 genindeki mutasyonlar neden olur .
Tanıtım
Smith-Magenis sendromu tıp literatüründe ilk kez 1982 yılında genetik danışmanı Ann Smith ve meslektaşları tarafından rapor edilmiştir. 1986'da Smith ve Dr. R. Ellen Magenis, sendromu daha da tanımlayan bozukluğu olan dokuz hastayı tanımladı. O zamandan beri, doktorların / klinisyenlerin bu karmaşık nörogelişimsel bozukluk ( NDD ) hakkında daha iyi bir anlayış geliştirmelerini sağlayan çok sayıda ek vaka tanımlanmıştır.
İşaretler ve Belirtiler
Smith-Magenis sendromu oldukça değişken bir hastalıktır. Mevcut spesifik semptomlar ve bozukluğun genel şiddeti bir kişiden diğerine değişebilir. Etkilenen bireylerin aşağıda tartışılan semptomların tümüne sahip olmayacağını ve her bir vakanın benzersiz olduğunu anlamak önemlidir. Ebeveynler, çocuklarının özel durumu, ilişkili semptomlar ve genel prognoz hakkında doktor ve tıbbi ekiple konuşmalıdır.
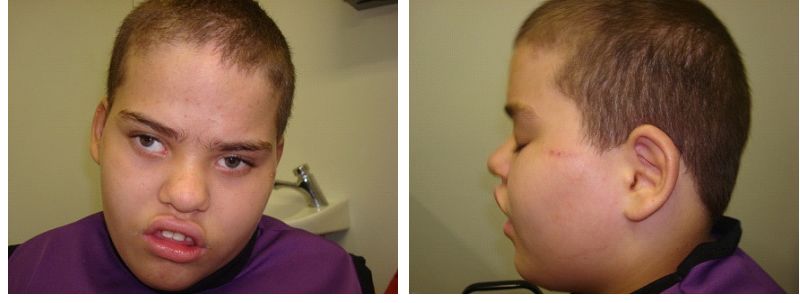
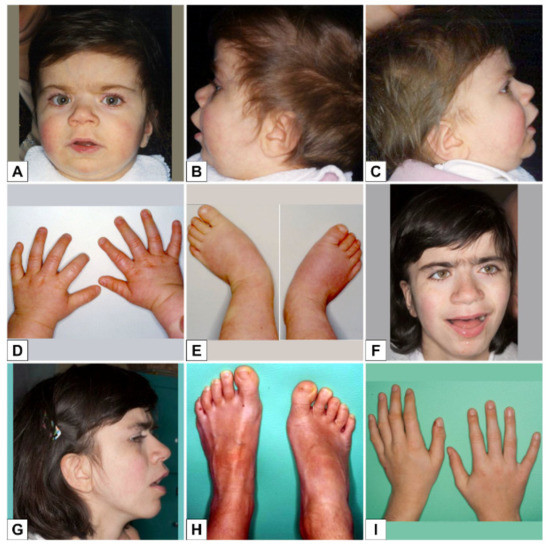
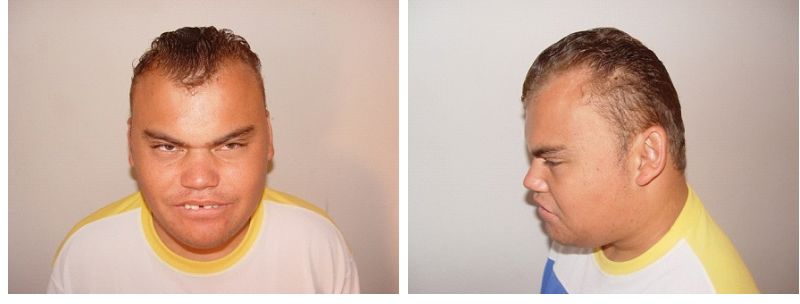
SMS'li birçok kişi, geniş, kare şekilli bir yüz görünümü, belirgin bir alın, normalden daha uzakta olan derin yerleşimli gözler ( hipertelorizm ), yukarı doğru eğimli bir palpebral ( göz ) çatlakları, geniş bir burun köprüsü gibi ayırt edici yüz özelliklerine sahiptir. tek bir uzun kaş ( sinophrys ), aşağı dönük ( dışa dönük; aşk tanrısı ) üst dudak, kısa, tam uçlu bir burun ve yüzün orta kısmının az gelişmişliği ( orta yüz retrüzyonu ) gibi görünecek şekilde kaşlar arasında büyüme. Kafa orantısız olarak kısa görünebilir ( brakisefali ). Etkilenen bazı bebekler anormal derecede küçük bir çeneye ( mikrognati ) sahip olabilir ve yüz görünümü pembe yanaklarla daha “kerubik”tir. Etkilenen bireyler yaşlandıkça, mikrognati değişebilir, böylece alt çene anormal şekilde dışa doğru çıkıntı yapar ( göreceli prognati ). Genel olarak, SMS ile ilişkili ayırt edici yüz özellikleri yaşla birlikte ilerler. Etkilenen bireyler ayrıca ikincil ( kalıcı ) dişlerin, özellikle küçük azı dişlerinin yokluğu ( agenesis ) ve pulpa odalarının genişlemesi ve diş köklerinin küçülmesi ile karakterize edilen bir durum olan taurodauntizm sergileyebilir; büyük dil ( makroglossia ) ve bruksizm ( diş gıcırdatma ) öyküsü ile açık kapanış da yaygındır.
Bebekler genellikle azalmış kas tonusuna ( hipotoni ), zayıf reflekslere ( hiporefleksi ) ve gelişme geriliğine katkıda bulunabilecek zayıf emme yeteneği gibi beslenme güçlüklerine sahiptir. Gelişememe, yaşa ve cinsiyete bağlı olarak beklenen oranda büyüme ve kilo alamama olarak tanımlanır. Bebekler genellikle sessizdir ve nadiren ağlama ve belirgin erken ifade edici konuşma gecikmesini yansıtan azalmış seslendirmeler ile kayıtsızdır. Ek olarak, etkilenen bebekler uzun süre şekerleme yapabilir ve genel gündüz uyuşukluk sergileyebilir. Gastroözofageal reflü bebeklik döneminde de yaygındır.
SMS'li bireyler değişen derecelerde bilişsel yeteneğe sahiptir. Birçok kişi hafif ila orta derecede zihinsel engellilik sergiler. Etkilenen bireyler genellikle konuşma ve motor becerileri edinmede ve gelişimsel dönüm noktalarına ulaşmada gecikmeler ( gelişimsel gecikmeler ) sergilerler. İfade edici dil, genellikle alıcı dil becerilerinden daha gecikir.
SMS'li çocuklarda belirli davranış sorunları ( uyumsuz davranışlar ) ortaya çıkar. Yaygın bir ilk işaret, erken çocukluk döneminde baş vurmadır. Sıklıkla "kendi kendine sarılma" olarak tanımlanan üst vücut sıkışmaları da yaygındır. Etkilenen çocuklar ayrıca dürtüsellik, hiperaktivite ve dikkat eksikliği bozukluğu, sık ve uzun süreli öfke nöbetleri, ani ruh hali değişiklikleri, tuvalet eğitimi güçlükleri, itaatsizlik ve saldırgan veya dikkat arayan davranışlar sergileyebilir. Kafa vurmaya ek olarak, etkilenen çocuklar el ısırma, yüze tokat atma, deri yolma ve bilek ısırma gibi kendine zarar veren başka davranışlar da geliştirebilirler. Tekrarlanan kafa çarpması potansiyel olarak retinanın ayrılmasına neden olabilir ve bu endişe verici olsa da yüksek risk değildir. Daha büyük çocuklar el ve ayak tırnaklarını çekebilir ( onikotillomani ) veya vücut açıklıklarına nesneler sokabilir ( poliembolokoilamania ). Etkilenen çocuklar heyecanlanma eğilimindedir ve dikkatleri kolayca dağılır. Davranış sorunları yaygın olmasına rağmen, birçok kişi, büyük mizah anlayışları ve yüzler, yerler ve şeyler için kolay uzun süreli hafıza ile sevimli ve ilgi çekici kişiliklere sahip olma eğilimindedir.
Etkilenen çocuklar, tekrarlayan orta kulak enfeksiyonları ( otitis media ) dahil olmak üzere kronik kulak enfeksiyonları yaşayabilir. İşitme kaybı çok yaygındır, tipik olarak hafiften hafif dereceye kadar değişir ve yaşla birlikte dalgalı ve ilerleyici bir işitme kaybı paterni gösterir. Hem iletken hem de sensörinöral işitme kaybı gelişebilir. İletim tipi işitme kaybı en çok erken çocukluk döneminde ( 10 yaş altı ) görülürken, sensörinöral işitme kaybı daha ileri yaşlarda ( 11 yaş – yetişkinlik ) daha sık görülür. İletim tipi işitme kaybı, ses dalgaları dış veya orta kulaktan iç kulağa uygunsuz bir şekilde iletildiğinde ortaya çıkar ve bu da sese duyarlılığın azalmasına neden olur. Sensörinöral işitme kaybı, iç kulakta ( koklea ) veya iç kulaktan beyne giden sinir yolunda hasar olduğunda gelişir. Etkilenen bazı çocuklar, belirli seslere veya frekanslara ( hiperakuzi ) anormal derecede duyarlı olabilir. Sık sinüs enfeksiyonları ( sinüzit ) de yaygındır. İlerleyici miyopluk ( miyopi ), şaşılık ( şaşılık ) ve korneanın olağandışı küçüklüğü ( mikrokornea ) gibi göz anormallikleri de oluşabilir.
Etkilenen çocuklar genellikle gırtlak ( ses kutusu ) veya çevresindeki dokuyu etkileyen anormalliklere sahiptir. Laringeal anormallikler, polip ve nodül oluşumunu veya sıvı tutulmasına ( ödem ) bağlı şişmeyi içerir. Ses tellerinin felci de gelişmiştir. Etkilenen çocuklar, konuşma sırasında ağzın yumuşak damağının düzgün kapanmadığı velofaringeal yetmezlik yaşayabilir. Etkilenen bireylerin dudak, dil ve çene kaslarını kontrol etmekte zorlandıkları oral sensorimotor disfonksiyon da gelişebilir ve dil çıkıntısına ve sık salya akmasına neden olabilir. Bu tür anormallikler nedeniyle, çocuklarda boğuk, derin bir ses gelişebilir. Bu anormallikler ayrıca konuşma gelişimindeki gecikmelere de katkıda bulunur.
Aşırı kilo alımı ve obezite, ergenlik döneminde görülebilir ve çocukların yaklaşık %90'ı 14 yaşına kadar aşırı kilolu veya obez olabilir. Etkilenen bireyler, boyları tipik olarak yetişkinlerde normal aralıkta olmasına rağmen çocukluk döneminde kısa boy sergileyebilir. Çocukların yaklaşık %50'sinin kanında alışılmadık derecede yüksek kolesterol seviyeleri olabilir ( hiperkolesterolemi ). Kronik kabızlık da sık görülen bir komplikasyondur.
Etkilenen bireylerde ortaya çıkan uyku bozukluğu, yaşam boyu süren kronik bir sorundur. Bebeklik dönemindeki uyku sorunlarına ( genel uyuşukluk ve “çok uykulu” ) ek olarak, etkilenen bireyler erken çocukluk döneminden ergenlik ve yetişkinliğe kadar devam eden önemli uyku bozuklukları geliştirir. Uyku döngüsü, uykuya dalma güçlüğü, kısa uyku döngüleri, REM uykusuna girememe ve gece ve sabah erken saatlerde ( 5:30 - 6:30 ) sık sık uyanma gibi sorunlarla karakterizedir. Genel olarak, uyku saatleri yaşa göre beklenenden daha azdır. Bozulan gece uyku döngüsünün bir sonucu olarak, etkilenen bireyler, gün içinde aşırı gündüz uyku hali veya uyku borcu olarak bilinen ve kronik bir sorun olmaya devam eden uyuşukluk dönemleri sergileyebilir. Uyku anormallikleri, incelenen vakaların %90'ından fazlasında rapor edilen, melatoninin ters çevrilmiş sirkadiyen ritmi ile ilişkilidir. Sirkadiyen ritim uyku bozukluğu, bir kişinin biyolojik saatleri normal 24 saatlik bir gün ile senkronize olmadığında ortaya çıkar. Spesifik olarak, normal olarak oluşan bir hormon olan melatonin yükselir ve düşer; yükselir, geceleri pik yapar ve uyuşukluğa neden olur. Melatonin seviyeleri sabahları azalır ve gün ortasında en düşük seviyelerine ulaşır. Ters sirkadiyen ritmi olan bireylerde melatonin seviyelerinin yükselmesi ve düşmesi tersine çevrilir. Gündüz yüksekleri geceleri pik yapar ve uyuşukluğa neden olur.
İskelet malformasyonları SMS'li bireylerde yaygındır ve omurganın önden arkaya eğriliğini ( lordoz ), omurganın hafif - orta yan eğriliğini ( skolyoz ), anormal derecede küçük eller ve ayakları ve belirgin şekilde düz veya çok kavisli olağandışı bir yürüyüş şekline neden olabilen ayaklar ( anormal geniş tabanlı yürüyüş ) olabilir. Nadir durumlarda, etkilenen çocukların vertebral anomalileri ve önkol ve dirsek sınırlamaları vardır.
Daha az sıklıkla, SMS'li bireylerde bağışıklık sistemi disfonksiyonu, tiroid fonksiyon anormallikleri ( hipotiroidizm ), kalp ( kalp ) kusurları, böbrek ( böbrek ) ve / veya idrar yolu malformasyonları, yarık dudak ve yarık damak ve nöbetler. Nöbet aktivitesi, nöbetin fark edilmeden gitmesi için gizlice ortaya çıkabilir ( subklinik nöbetler ). Periferik sinir sisteminin herhangi bir bozukluğu için genel bir terim olan periferik nöropati de ortaya çıkabilir. Periferik nöropati, öncelikle merkezi sinir sistemi dışındaki sinirleri ( yani beyin ve omurilik ) etkileyen herhangi bir bozukluğu kapsar. Semptomlar, SMS'de yaygın olarak görülen ağrıya duyarlılığın azalmasını içerebilir. Periferik nöropati genellikle duyu kaybı veya karıncalanma, yanma gibi anormal duyumlar ile ilişkilidir.
Nedenler
Etkilenen bireylerin yaklaşık %90'ında, kromozom 17'nin ( 17q11.2 ) kısa kolunun ( p ) bir kısmı eksiktir ve bu, silinmiş veya monozomik olarak adlandırılır. İnsan hücrelerinin çekirdeğinde bulunan kromozomlar, her birey için genetik bilgiyi taşır. İnsan vücut hücreleri normalde 46 kromozoma sahiptir. İnsan kromozom çiftleri 1'den 22'ye kadar numaralandırılmıştır ve cinsiyet kromozomları X ve Y olarak adlandırılmıştır. Erkeklerde bir X ve bir Y kromozomu ve dişilerde iki X kromozomu vardır. Her kromozomun “p” olarak adlandırılan kısa bir kolu ve “q” olarak adlandırılan uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, "kromozom 17p11.2", kromozom 17'nin kısa kolundaki 11.2 bandına atıfta bulunur. Numaralandırılmış bantlar, her kromozomda bulunan binlerce genin yerini belirtir.
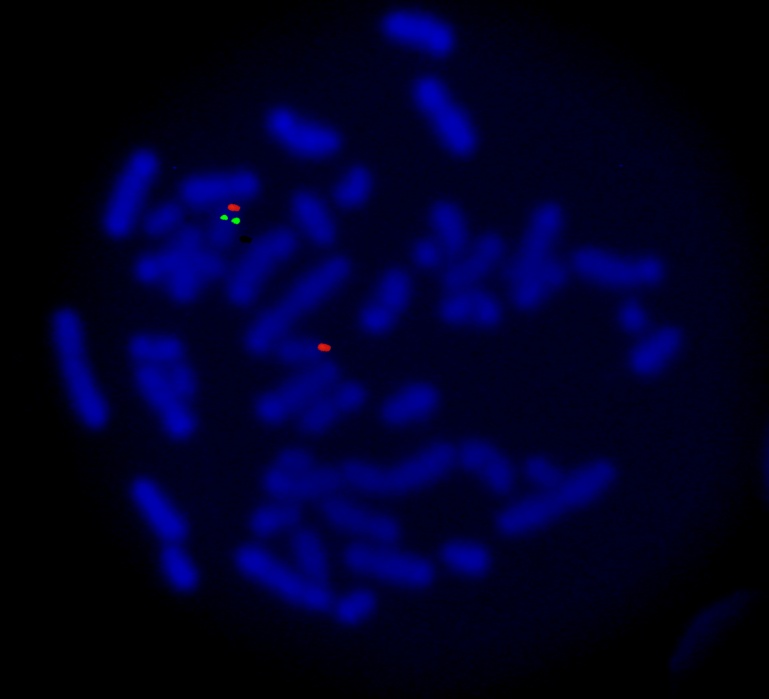
SMS'li bireylerde, kromozom 17'nin silinen bölümü, retinoik asit ile indüklenen 1 ( RAI1 ) genini içerir. Genler, vücudun birçok işlevinde kritik bir rol oynayan proteinlerin oluşturulması için talimatlar sağlar. Bir monozomik kromozom anormalliği nedeniyle bir gen eksik olduğunda, o genin protein ürünü azalır. Belirli bir proteinin işlevlerine bağlı olarak, bu, beyin de dahil olmak üzere vücudun birçok organ sistemini etkileyebilir. RAI1 geni tarafından üretilen ( kodlanan ) proteinin spesifik işlevleri tam olarak anlaşılamamıştır.
SMS'deki kromozomal değişikliğin kesin nedeni bilinmemektedir. Tıp literatürü, belgelenen neredeyse tüm vakaların, bilinmeyen nedenlerle meydana gelen spontane ( de novo ) bir genetik değişimden kaynaklandığını göstermiştir.
Nadir durumlarda, SMS, ebeveynlerden birinde kromozomal dengeli bir translokasyon nedeniyle çok erken embriyonik gelişim sırasında bir hatanın sonucudur. İki veya daha fazla kromozomun parçaları kırılırsa ve yer değiştirirse, değiştirilmiş ancak dengeli bir kromozom seti oluşturursa, bir translokasyon dengelenir. Bir kromozomal yeniden düzenleme dengeliyse, genellikle taşıyıcıya zararsızdır. Bununla birlikte, taşıyıcının yavrularında daha yüksek anormal kromozomal gelişim riski ile ilişkili olabilirler. Bu durumlarda, çocukların klinik özellikleri, 17 dışındaki diğer kromozomların ek dengesizliklerinden etkilenebilir. Kromozomal testler, bir ebeveynin dengeli bir translokasyona sahip olup olmadığını belirleyebilir. SMS'li çocuğu olan ve kromozom analizi normal olan anne babalarda ileriki gebeliklerde tekrarlama riski %1'in altındadır.
SMS vakalarının geri kalan %10'una RAI1 genindeki mutasyonlar neden olur. Bu mutasyonlar, aile öyküsü olmadan ( yani yeni mutasyon ) rastgele meydana gelebilir veya otozomal dominant bir şekilde kalıtsal olabilir. Genetik hastalıklar, baba ve anneden alınan kromozomlarda bulunan belirli bir özellik için genlerin kombinasyonu ile belirlenir. Baskın genetik bozukluklar, hastalığın ortaya çıkması için anormal bir genin yalnızca tek bir kopyası gerektiğinde ortaya çıkar. Anormal gen, ebeveynden kalıtsal olabilir veya etkilenen bireyde yeni bir mutasyonun ( gen değişikliği ) sonucu olabilir. Anormal genin etkilenen ebeveynden yavruya geçme riski, ortaya çıkan çocuğun cinsiyetine bakılmaksızın her hamilelik için %50'dir. RAI1'deki mutasyonlargen, normal olarak gen tarafından üretilen protein ürününün yetersiz fonksiyonel kopya seviyelerine yol açar.
Tıbbi literatürde bildirilen iki ailede, germ hattı mozaikliği nedeniyle SMS meydana geldi. Germ hattı mozaisizminde, bir ebeveynin üreme ( germ ) hücrelerinin bir kısmı RAI1'i taşır. Gen mutasyonu veya kromozom 17p delesyonu, diğer germ hücrelerinde görülmez ( mozaiklik ). Ek olarak, bir ebeveynin diğer hücrelerinde de bu kromozomal anormalliklerin hiçbiri yoktur; sonuç olarak, ebeveynler etkilenmez. Bununla birlikte, sonuç olarak, ebeveynin çocuklarından biri veya daha fazlası, kromozomal bir anormallik ile germ hücresini miras alabilir ve bu da SMS'nin gelişmesine yol açar. Görünüşte etkilenmemiş ebeveynlerin bu bozukluğa sahip birden fazla çocuğu olduğunda germ hattı mozaisizminden şüphelenilir. Bir ebeveynin mozaik germ hattı kromozomal anormalliğini bir çocuğa geçirme olasılığı, ebeveynin anormalliği olan germ hücrelerinin yüzdesine karşı olmayan yüzdesine bağlıdır. Hamilelikten önce germ hattı mutasyonu veya kromozom anormalliği için herhangi bir test yoktur.
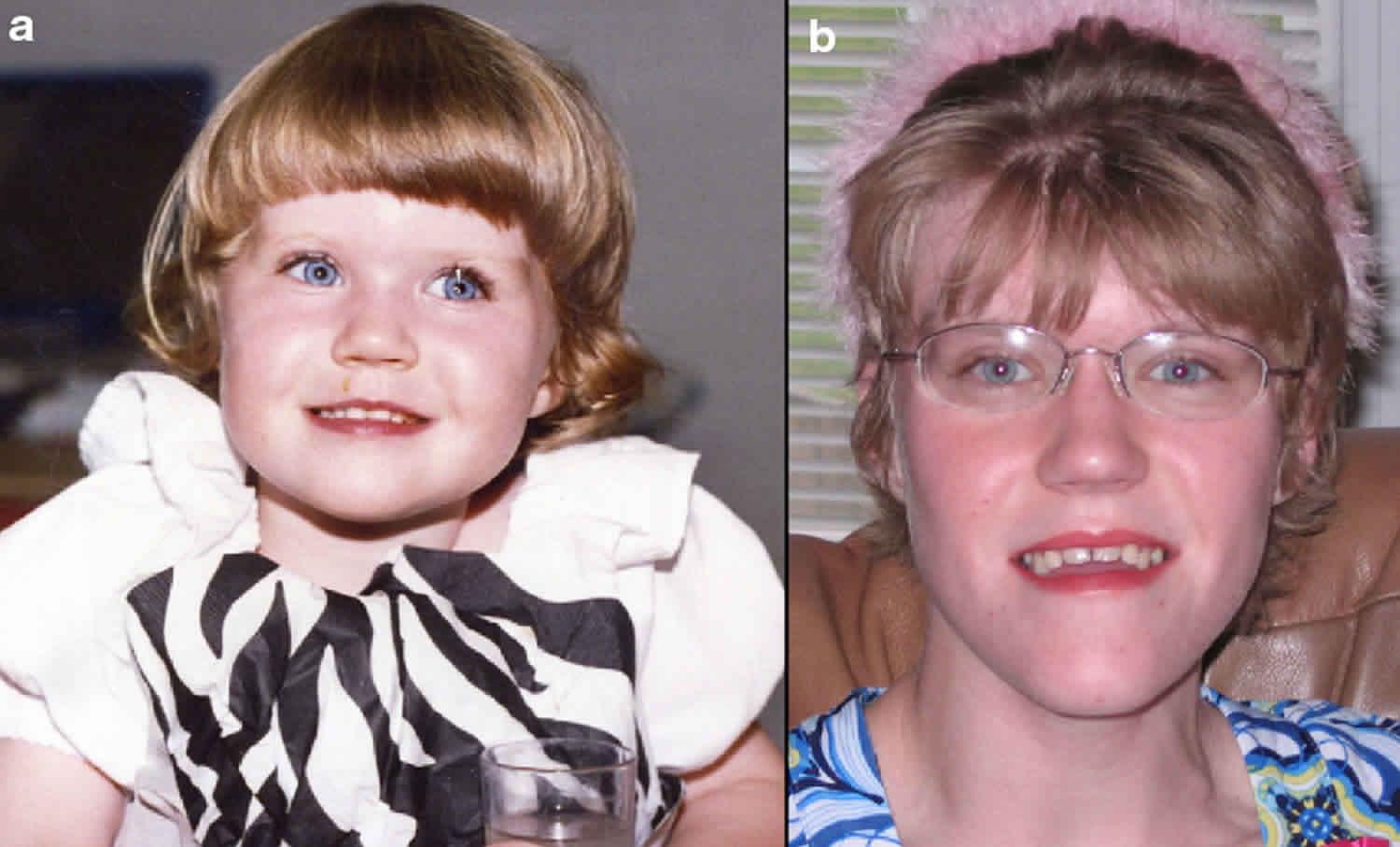
SMS'li bir bireyden doğan bir çocuk, bozukluğa neden olan delesyon veya RAI1 mutasyonunu kalıtım yoluyla almak için teorik olarak %50 risk altındadır. Genel olarak SMS'deki doğurganlık tam olarak anlaşılmamıştır; ancak tıbbi literatürde SMS'li bir annenin SMS'li çocuğu olduğuna dair en az bir rapor bulunmaktadır.
Etkilenen Popülasyonlar
Smith-Magenis sendromu erkekleri ve kadınları eşit sayıda etkiler. Amerika Birleşik Devletleri'ndeki genel popülasyonda görülme sıklığının 15.000 - 25.000 kişide 1 olduğu tahmin edilmektedir. Bununla birlikte, vakalar teşhis edilemeyebilir veya yanlış teşhis edilebilir, bu da genel popülasyonda SMS'nin gerçek sıklığını belirlemeyi zorlaştırır. SMS tüm dünyada ve tüm etnik gruplarda bildirilmiştir.
İlgili Bozukluklar
Aşağıdaki bozuklukların semptomları Smith-Magenis sendromunun semptomlarına benzer olabilir. Karşılaştırmalar ayırıcı tanı için faydalı olabilir.
SMS'de görülenlere benzer belirti ve semptomlara sahip birçok farklı kromozomal bozukluk vardır. Bu tür bozukluklar arasında Down sendromu, Williams sendromu, Prader-Willi sendromu, Angelman sendromu, Sotos sendromu, frajil X sendromu, kromozom 22q11.2 delesyon sendromu, 9q34 delesyon sendromu ( Kleefstra sendromu ), 2q37 delesyon sendromu, 2q23.1 delesyon sendromu ve 1p36 silme sendromu. SMS'li bireyler genellikle başlangıçta obsesif-kompulsif bozukluk, yaygın gelişimsel bozukluk veya dikkat eksikliği hiperaktivite bozukluğu gibi bir psikiyatrik tanı alırlar. SMS'li bazı çocuklara başlangıçta bir otizm spektrum bozukluğu teşhisi konur.
Teşhis
Smith-Magenis sendromunun teşhisi, karakteristik semptomların tanımlanmasına, ayrıntılı bir hasta ve aile öyküsüne, kapsamlı bir klinik değerlendirmeye ve çeşitli özel genetik testlere dayanır. Silme 17p11.2 ( sitogenetik analiz veya mikrodizi ) veya RAI1 gen mutasyonu tanımlandığında SMS teşhisi doğrulanır.
Klinik Test ve Çalışma
Geçmişte, G-bandı analizi olarak bilinen ve 17p kromozomu üzerinde eksik ( silinmiş ) materyali gösteren spesifik bir kromozomal çalışma, SMS teşhisinin elde edilmesine yardımcı olmak için kullanılıyordu. Kromozomlar bir kan örneğinden elde edilebilir. Bu test sırasında kromozomlar daha kolay görülebilmeleri için boyanır ve daha sonra 17p kromozomunun eksik parçasının tespit edilebileceği ( karyotipleme ) bir mikroskop altında incelenir. Kesin kırılma noktasını belirlemek için floresan in situ hibridizasyon ( FISH ) olarak bilinen daha hassas bir test gerekli olabilir. Bir FISH muayenesi sırasında, belirli bir floresan boya rengiyle işaretlenmiş problar, belirli bir kromozoma bağlanır ve araştırmacıların kromozomun o belirli bölgesini daha iyi görmelerini sağlar.
Kromozomal mikrodizi analizi olarak bilinen daha yeni bir teknik de kullanılabilir. Bu muayene sırasında, bir kişinin DNA'sı, kromozomal anormalliği olmayan bir kişinin ( 'kontrol' kişisi ) DNA'sı ile karşılaştırılır. DNA örnekleri arasında bir fark bulunduğunda bir kromozom anormalliği not edilir. Kromozomal mikrodizi analizi, çok küçük değişikliklerin ( eksik veya kopyalanmış segmentler ) veya değişikliklerin tespit edilmesini sağlar.
Moleküler genetik testler, bir RAI1 gen mutasyonu nedeniyle SMS olduğundan şüphelenilen kişilerde tanıyı doğrulayabilir. Moleküler genetik testler, belirli durumlarda SMS'e neden olduğu bilinen RAI1 genindeki mutasyonları tespit edebilir , ancak yalnızca özel laboratuvarlarda tanı hizmeti olarak kullanılabilir.
Standart Terapiler
Tedavi
Tedavi, bir uzman ekibinin koordineli çabalarını gerektirebilir. Çocuk doktorları, cerrahlar, kardiyologlar, diş uzmanları, konuşma patologları, odyologlar, oftalmologlar, psikologlar ve diğer sağlık profesyonellerinin çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlamaları ve etkilemeleri gerekebilir. Genetik danışmanlık, etkilenen bireyler ve aileleri için faydalıdır. Tüm aile için psikososyal destek de önemlidir.

Tedavi semptomatik ve destekleyicidir. Etkilenen çocukların en yüksek potansiyellerine ulaşmalarını sağlamak için erken müdahale önemlidir. Yararlı olabilecek hizmetler arasında, çocuğun duyusal uyaranlara tepkisini düzenlemeye yardımcı olmak için belirli duyusal etkinliklerin gerçekleştirildiği özel iyileştirici eğitim, konuşma / dil terapisi, fizik tedavi, uğraşı terapisi ve duyu bütünleme terapisi yer alır. Uygun olduğunda ek tıbbi, sosyal ve mesleki hizmetler önerilebilir.
Dikkat eksikliği veya hiperaktivite gibi davranış problemlerini tedavi etmek için bazı ilaçlar kullanılabilir. Potansiyel olarak SMS ile ilişkili uyku bozukluklarını tedavi etmek için özel ilaçlar da kullanılmıştır. Melatonin seviyelerini normalleştirmek için yatmadan önce alınan melatonin takviyesi, anekdot raporlarında fayda göstermiştir. Bir Fransız çalışmasında gündüz melatonin salgısını inhibe etmek / bastırmak için sabahları B-bloker asebutolol kullanımı bazı faydalar göstermiştir.
Beslenme zorlukları, tanımlama ve uygun tedavi gerektirir. Ek tedavi, spesifik semptom için standart yönergeleri takip eder. Örneğin, nöbetleri tedavi etmek için nöbet önleyici ilaçlar ( anti-konvülzanlar ) kullanılabilir.
Prognoz
SMS'nin oldukça değişken doğası nedeniyle, bireysel vakalar için prognoz hakkında genelleme yapmak mümkün değildir. Etkilenen bazı kişiler, ailelerinin ve arkadaşlarının desteğiyle iş sahibi olabilmiş ve hatta yarı bağımsız olarak yaşayabilmiştir. Bununla birlikte, diğerleri sürekli bakım gerektirir ve aile ile veya bir konut tesisinde yaşamaları gerekebilir. Yukarıda belirtildiği gibi, ebeveynler, çocuklarının özel durumu ve genel prognozu hakkında doktor ve tıbbi ekiple konuşmalıdır.